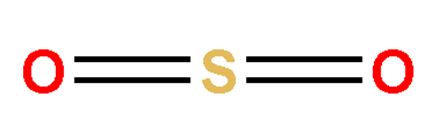Diatomic oxygen(O2)
From a statistical point of view, oxygen is the sports superstar of the elements. It is the only element to appear in the top 3 by weight percentage. It’s the third most abundant element in the universe; second on earth and life’s first. There are life forms that can get by without diatomic oxygen, but all life depends on the organic compounds and water that can only be formed with oxygen atoms. Here we will begin with the gas that no land animal or plant can do without.
I have opened my share of dry cells and alkaline batteries to reveal their inner workings to myself, and then eventually to students. After sawing off the batteries’ top, there is a characteristic smell. It has reminded me of my first childhood attempt at making the essential element oxygen from my parents’ hydrogen peroxide (H2O2) and an ingredient common to both zinc and the old ammonium-based batteries: manganese dioxide. This fine black powder catalyzes the chemical breakdown of each pair of hydrogen peroxide molecules into a pair of water molecules and one diatomic molecule of oxygen.
You can check for oxygen’s odorless presence with a glowing wood splint. If the concentration in a test tube is high enough, you not only see the splint characteristically burst into a flame, you even get a nice popping sound. In simpler reactions, regardless of whether oxygen interacts with metals or non-metals, the compounds have less potential energy than the reactants. Since energy is conserved, the excess energy is liberated in the form of light and/or heat. But for living aerobic cells, some of the energy from oxidized food molecules is invested in making other compounds which eventually facilitate non-spontaneous reactions. Instead of wreaking havoc like a forest fire after a lightning storm, the subsequent anabolic reactions decrease the internal entropy of the organism, sustaining it. We often hear that oxygen “burns” food. In comparing cellular respiration to combustion, there are indeed common starting and end- products, but the former has other intermediates—a whole other story in between, a difference between life and destruction.
Why do biological food molecules exist? Life can use energy sources to build larger molecules. Later, it evolved photosynthesis, a way to use light as its energy source. In doing so, electrons are emitted from its pigment-centers, solar cells far more sophisticated than the ones we have invented. These electrons help bond smaller molecules into glucose. But that would not in itself be energetically feasible unless the photosynthetic organism could tap into the bulk of the solar energy, which is not contained in the electrons.
Oxygen enters the picture. About 3 billion years ago, in prokaryotes, photosynthesis not only involved electrons leaving pigments, but in order to make pigments available for excitation again, electrons had to return to them. Each of the four electrons, delivered one at a time, came from a pair of water molecules that dissociated to also produce 4 positive ions of hydrogen and a molecule of oxygen. The oxygen slowly filled the atmosphere, which paved the way for the evolution of animals. The charged particles concentrated on one side of a membrane, storing potential energy and allowing the bulk of the energy to be used for the so-called dark reactions of photosynthesis.

The oxygen-evolving-complex (OEC) ( Figure 1) is an enzyme with an imbedded cluster of Mn4O5Ca. Notice how manganese, the atom and battery-ingredient involved in the catalytic breakdown of hydrogen peroxide makes another appearance. Sitting in one of the pigment-systems of plants and algae, OEC is the precise location of where water molecules break up, deliver their electrons to chlorophyll and yield acid and oxygen gas. But how the enzyme exactly operates is still a big biochemical mystery. It’s certainly more sophisticated than the way we electrolyze water into hydrogen gas and oxygen.
i. Oxygen and fire
It’s hard to discuss oxygen without remembering that along with fuel and heat, it is one of the three parts of the fire triangle. Remove any of the trio and the fire is extinguished. Let’s explore the chemistry of fire and oxygen’s role by examining campfires.


Figure 2. Fire is an interaction of energy and matter, including free radicals and diatomic carbon. On the right, birch bark. Pictures by author.
Birch bark, which is found in many parts of the U.S., Canada, Europe and China, is a great way to start a campfire. Rich in terpenoids, the paper-thin material ignites easily. The heat it releases provides enough activation energy to set small twigs ablaze, which of course should be placed in a tee-pee arrangement to let in more oxygen. All of this should take place in a pit surrounded by stones, not to let wind take heat away from the young fire and not to burn the forest down.
The hues of a flame are rarely constant for a second, a hint that something complex is occurring within them. There is a set of chain mechanisms involving intermediate molecules that are needed for subsequent steps. Many of the in-between products are radicals, reactive molecules with unpaired electrons. Radicals are often created in the high temperature regions of the flame, but they diffuse back into the colder regions where they are needed to generate the final products. To reveal more details, laser-based investigative techniques have been used so as not to disturb the flame. Even when burning as simple a molecule as diatomic hydrogen, radicals like O, H, OH and HO2 form. When the combustion of hydrocarbons like cellulose and lignin in wood takes place, we get a greater variety of radicals, some of them carbon-centered. C2 and the radical CH arise in excited form and release blue and green light. Lignin-derived radicals involving benzene structures are not the healthiest things to inhale, but mere occasional exposure is probably nothing to worry about, unless there’s a concentration of wood-burning stoves in an area. All of this underlines the fact that whenever we write an overall equation for a fuel consumed in a fire, it’s like we are seeing only the ingredients of a recipe and the final product without witnessing the cooking.
Hot, gaseous products of combustion expand and rise, stretching flames vertically. The ascension leads to pressure gradients, and fresh air is pushed into the fire. The circulation supplies it with more oxygen, the electron-thief that campfires depend on to release heat as more tightly bonded products like water and carbon dioxide are created. There’s energy needed to drive molecular fragments of cellulose apart, in the same way that you need to exert force against gravity if you want to push a ball up a hill. But once at the top, the ball can roll further down on the other side. With chemicals, it’s not the combination of mass, gravity and varying heights that accounts for differences in potential energy but Coulombic forces acting over a variety of distances between positive atomic nuclei and valence electrons.
Why is a wood flame predominantly yellow orange? It has been proposed that it’s not the result of electron transitions; what’s responsible is the incandescence of particles at about 1100 to 1200 oC. Since the combustion of wood is incomplete, the flame’s soot particles, some of which are elemental carbon (others are polymers), emit part of their vibrational energy as photons. How fast the molecules vibrate depends on their temperature, and the hotter the surface, the higher the frequency of the photons emitted. The same mechanism accounts for the red glow of logs at the base of a fire. But the temperature is lower, about 700 to 1000oC, hence a color of a longer wavelength and a lower frequency. Different parts of the charcoal emit light of slightly different frequencies, intermittently and in different directions. A point on the surface of the charcoal particle that has just emitted photons will have lost energy and cooled slightly. Although exothermic reactions quickly compensate, from that same spot, the temperature will not necessarily be identical, especially considering air movement and the exact frequency of photons is not necessarily replicated.
It’s well known that the ease of ignition and burning rate of wood vary greatly with moisture content. Specifically, a 10% drop in moisture-content results in an increase of 20–30% in burning rate. When wood is too dry the combustion rate increases, but an inadequate oxygen supply leads to more undesirable emissions. The combustion rate also depends on boundary conditions and the species being burnt. Why does it vary with tree type? Wood composition is not constant. Wood is essentially a matrix of cellulose and other carbohydrate fibers (hemi cellulose) reinforced by the adhesive binding action of lignin. But hardwoods can have anywhere from 18 to 25% lignin along with varying amounts of hemicellulose, usually a partly acetylated, acidic xylan. Softer woods have other hemicellulose fibers and more “binder”, 25 to 35% lignin. There is also an assortment of oils and secondary products present.
The different wood recipes not only affect kinetics but thermochemistry. Softwoods, compared to hardwoods, release on average an extra 5% of heat, a maximum of 21 instead of 20 MJ/kg, to be precise. From the point of view of carbon dioxide emissions, it’s not a good idea to rely on wood as a primary fuel. For every MJ of heat obtained from wood, on average, 80 g of CO2 are emitted. In contrast, natural gas combustion only puts out 50 g per MJ.
ii. A hands-on example of oxygen’s explosive reactivity
When we were preteens, someone in our neighborhood showed us how to make a tennis-ball cannon. It consisted of cutting off the top and bottom of about 6 cans of beer and then taping them together. For the seventh can—the cannon’s base and reaction chamber—we left the bottom intact and cut off only a circular part of the can’s top so it could act as a support for a tennis ball. About an inch from the bottom we also poked about a 1 cm- hole and then taped the base to the rest of the can assembly. Through the hole, we poured in a bit of gasoline, slid the ball in, shook the cannon, waited and then ignited it with a match.
The first time we fired the cannon, we were in the fields behind our street. Its range astonished us as the ball almost inadvertently struck a woman on a 2nd floor balcony, hundreds of feet away. She was putting out her laundry, and she hollered at us when the ball bounced off the brick wall behind her. A week later, just before we tried it again, an inquisitive adult by the name of Eduardo laughed in disbelief when we told him what we expected to happen. The cannon was almost in a vertical position when we fired it. Eduardo’s jaw dropped almost as low as his clavicle. The ball had gone so high that we momentarily lost sight of it.
After hearing the story, my students were exploding with enthusiasm to build their own. In the early years of my teaching career, beer can-diameters had already been made incompatible with the size of tennis balls. But the biggest problem was that it would have been irresponsible to use gasoline around students. The heat of combustion for its safer substitute, ethanol, was no match for that of gasoline. Launching it became a hit and miss affair, and we did not initially get the bang, or the range promised by my recollection.
Understanding the theory helped us out in the long run. Before ignition, some alcohol molecules are in the vapour phase. Along with the air molecules in the base of the cannon, they exert pressure on the ball. But that pressure is not significantly different from the air pressure exerted on the other side of the ball, facing the cannon barrel’s exit.
After igniting the alcohol, we have a different matter. If there’s sufficient oxygen, the alcohol can optimally react with oxygen gas in a 1 to 3 ratio to yield carbon dioxide and water in a 2 to 3 ratio. The total potential energy of the oxygen and alcohol lie mostly within the bond energies of oxygen and alcohol’s intramolecular bonds. That sum exceeds that of the intramolecular bonds of the products of combustion, which is why the excess energy is released.
The sudden release of heat excites the molecules of the CO2, H2O and of the part of the air that did not react. As these molecules pick up speed in a confined volume, they exert far more force per unit area on the ball. The pressure deforms the ball, and as it regains its shape, some of the energy becomes the ball’s kinetic energy, accelerating it out of the cannon. But the gas-ball collisions are not elastic. Some of the energy bunches up air molecules, forming waves. In other words, some of the energy ends up as a whistling or thunderous sound, depending on how much heat was released. With an unsealed ignition-hole and reliance on tape, some exhaust gases escape, wasting even more energy.

One year I was in my brother-in-law’s steel workshop when I noticed some spare metal cylinders. I got him to weld one to a base, told him where I wanted the hole, and suddenly we had a much more solid tennis ball cannon. When I brought it to school, to get more of the alcohol vapours to ignite, to approach a more stoichiometric ratio, we decided to use a portable oxygen tank to inject gas into the chamber. To prevent it from escaping, we sealed the hole with plasticine through which we had inserted a firecracker fuse.
The firing of the cannon was spectacular(Figure 3), judging from the exploding sound and voices after the fuse went off. The ball zoomed across the parking lot. No one saw the ball in flight, but we found it because some of us heard it hit a tree that intercepted its trajectory. Here it is in action:
There’s more than enough muzzle velocity for the ball to detach someone’s retina, so safety goggles are a must. But that danger aside, it would be nice if gun users would some day replace their bullets and guns with alcohol-cannon projects.
Ozone(O3)
In various classroom demonstrations,I often used a Tesla coil. The one we owned looked like a giant pencil. Its removable tip was pointy and metallic. When plugged into an alternating current source, it would generate a high voltage and low current that could dimly illuminate an unconnected fluorescent tube without making contact. In between the tip of the coil and the pair of the bulb’s contacts, there would be a blueish arc, resembling a mini bolt of lightning. There would also be a faint, unpleasant chlorine-like smell that students sitting in the front row would detect. What was going on?
The blue color is already an indication that air molecules are being excited by the high voltage. But the smell results from the formation of ozone, O3 . The coil has enough energy to split each oxygen molecule into a pair of oxygen atoms, one of which then bonds to an unsplit molecule of O2 to yield O3.
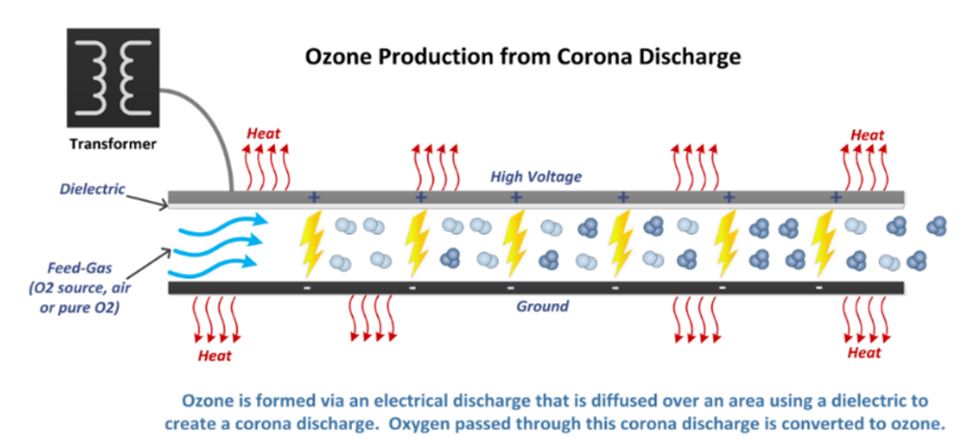
The commercial generation of ozone is always done on site because the molecule is unstable. A high voltage alternating current (600 to 20 000 volts) is applied across a dielectric discharge gap that contains either air or pure oxygen (Figure 4). (A dielectric is a ceramic material that keeps the charges separated.) Between the plates we observe a bluish corona, like the plasma we saw at the tip of Tesla coil when it approached a metallic surface, but on a larger scale.
At a water treatment plant, ozone is a friend. It is better than chlorine at killing harmful microorganisms without leaving chlorinated hydrocarbons behind. Ozone, unlike chlorine, is even effective at breaking down most pesticides. An ozone dose of 0.4 mg/L for only 4 minutes is effective for pre-treated water. In 2016, the city of Montreal treated about 800 liters of water per person daily, of which about 75% was treated with ozone. Although the city’s consumption habits have been improving with better designed flush toilets, less watering of lawns and replacement of old water mains, they are still wasteful compared to most European cities.
Ozone unfortunately breaks down faster than chlorine, so some of the latter is still used, in case drinking water is recontaminated between its source and destination. But use of ozone significantly lowers the concentration of chlorine needed, rendering the water healthier and almost odorless. Many cities also treat sewage with ozone in the final stages before releasing it into the rivers to reduce bacteria and simplify the soup of compounds that would otherwise be ingested by aquatic life and people downstream.
In more recent years there have been many commercial attempts at retarding fungal growth on transported fruits using very low levels of ozone. A 2016 review study concluded that it’s not clear how efficient the gas really is at reducing spoilage. In medicine an ozonized oil seems to be effective against dermatophytes that attack soles of the feet.
Outside of a controlled environment, ozone is undesirable at ground level. Like the chlorine whose smell it resembles, it is a strong oxidizing agent, attacking rubber, eyes and lungs. Once oxygen was introduced into atmosphere by photosynthesis, another event of life-induced chemical evolution occurred. Up in the stratosphere some of the ultraviolet light from the sun, UVC, acted like the Tesla coil, creating ozone from oxygen. Molecular oxygen continues to protect us from dangerous UVC, whose wavelengths are in the 100 to 200 nanometer regions(Figure 5).
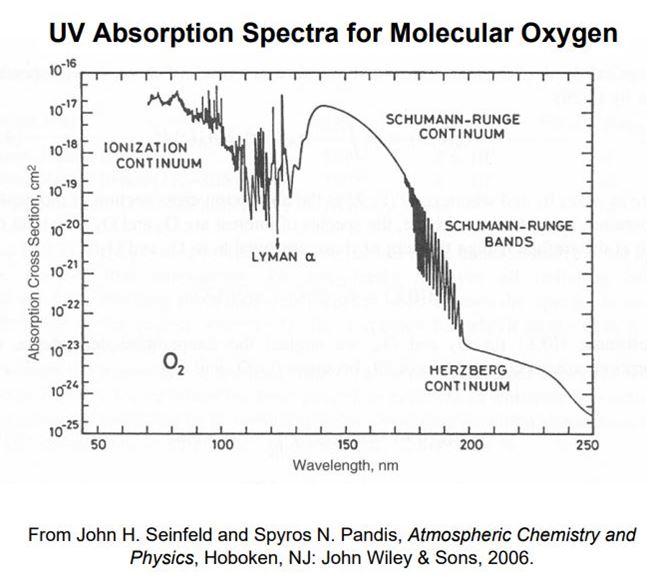
However, as Figure 5 indicates, molecular oxygen does not absorb well in the 200 to 280 nm region(UVB) of the ultraviolet spectrum. Ozone, instead, is capable of absorbing in that ultraviolet region (Figure 6). When each ozone molecule takes in energy of that wavelength it becomes one diatomic oxygen and one oxygen atom. Heat is released, keeping the stratosphere warmer than the troposphere underneath it but also providing atomic oxygen to keep the ozone layer at steady state.

Of course, we all know, thanks to the efforts of Rowland and Molina, that the steady state has been threatened, especially over Antarctica. Polar stratospheric clouds trap NO2 from volcanic emissions. That gas would normally neutralize the Cl from molecules like Freons which were common in refrigerating coolants. But once free, the Cl would compromise ozone concentrations by inducing its own steady state. How? Although the exact mechanism varies with latitude, basically the story goes like this. Cl is a neutral atom of chlorine with a valence of 7, a so-called radical because the 7th valence electron is unpaired. First the Cl radical plucks out an oxygen from O3 to create O2 and the radical ClO. Then ClO reacts with an ingredient of ozone-synthesis, atomic oxygen, to produce molecular oxygen and a Cl radical. The latter is now available to re-attack ozone, repeating the destructive cycle.
Hydroxide ion (OH–)

If you collect the white ashes from a wood fire, you will have a source of potassium carbonate, K2CO3. This water-soluble compound, when added to water, will create a solution that feels slippery between the thumb and the index. Why?
Water has an extremely small concentration of positive(H+)ions negative OH ions. These OH ions (OH–) called hydroxide have a negative one charge and without them we would not be able to dissociate water into oxygen gas. They are in equilibrium with unsplit water molecules and normally the positive and negative ions exist in equal numbers, keeping water’s pH neutral. While the negative-two carbonate ion uses up an H+ from the water to become hydrogen carbonate, the baking- soda ion that flows in blood and rivers, water is left with an excess of hydroxide. It has become a base or an alkaline solution. Some of those ions break up the oils of the skin into fatty acids, ingredients of soap—hence the slippery feel.
This is essentially how soap is manufactured on a large scale. Instead of skin oils they use vegetable fats like low-quality olive oil, palm and coconut oil. And instead of potassium carbonate, they use potassium hydroxide (KOH), which is a stronger base. Since the reaction also produces glycerin, salt is used to separate the two products. After removing the glycerin from the top, at this stage there is still some unreacted fat. An even more concentrated solution of hydroxide is added, and then additional steps are required to purify the soap. Finally, to make it more appealing, scents are added to the soap in one of the last stages of manufacturing. As one could see, it’s not just a one-shot deal that one imagines from seeing a chemical equation of the main reaction. The literal interpretation of the equation just leads to a mess in the lab!
To mention that potassium hydroxide is also used to make alkaline batteries, synthetic fertilizer and paper would be describing just the tip of the iceberg. It is a highly versatile compound. They once made it by reacting limewater with potassium carbonate. Nowadays, both sodium hydroxide (NaOH) and KOH are made by electrolyzing the respective aqueous chloride solutions. Chlorine and hydrogen gases are both byproducts of this reaction and are collected. Interestingly this is a side reaction if you try to electrolyze tap water, which has chlorides as impurities. You can always tell which students(or technicians and teachers!) are being honest in reporting their data. The expectation of a 2:1 ratio from the reaction of pure water skews many of the results. The side-reaction, in fact, creates a higher ratio because not only is it a second source of hydrogen, but more importantly, while the chloride ions compete with hydroxide to give up electrons, less oxygen gas is produced.
Rust (various oxides and hydroxides of iron)

I recall hearing about a woman who, decades ago, had a rust-prone Dodge model that was free of rust. It wasn’t because she kept the car locked up but because, unlike most people who buff their cars only in the summer when they need it the least, she waxed her Dodge a dozen times over the winter. In the overall reaction of atmospheric corrosion of iron, the reactants are iron, oxygen and water. Washing and drying prevents dirt from trapping moisture and washes out salt that accelerates corrosion. Waxing sets up a barrier between oxygen and iron.
The product of rust, contrary to what basic chemistry textbooks say, is more than just Fe2O3. The rust layer consists of a variety of minerals, including magnetite (mainly Fe3O4), maghemite(ϒ-Fe2O3), goethite, lepidocrocite and akaganeite ( mostly α, ϒ and β forms of FeOOH, respectively), reduced lepidocrocite (ϒ-FeOHOH) and ferrous hydroxide(Fe(OH)2). Such a minestrone is a hint that the chemistry is more complicated than previously imagined.
In the first stage of rusting, iron or low-alloy steel loses electrons and becomes soluble Fe2+. But the electrons are only partly taken up by oxygen. The majority are accepted by another ionic form of iron, Fe3+. Only after the latter are used up does oxygen pick up the slack. Along with water, oxygen picks up electrons to produce hydroxide which bond with Fe2+ to create a variety of iron compounds, which are then further oxidized. As the water from the electrolyte film evaporates, it slows down the whole process. Since humidity influences the rate of evaporation, it determines how many cycles of this process iron experiences over time.
Intermediate compounds with Fe3+ have low solubility. This slows down further formation of rust, but in the presence of sea salt or street salt, they are displaced by chloride ion. As Fe3+ ions go into solution, they are subject to a variety of reactions known as iron hydrolysis. Specifically, they react with water to form three different hydroxide complexes and, in each case, acidic ions. The acid then accelerates corrosion. Any acid already in the environment due to acidic precipitation will be even more efficient at producing rust.
There’s a connection between corrosion and the evolution of life. After the evolution of photosynthesis, almost all the O2 made by early cyanobacteria was used up in converting large amounts of Fe2+ in the oceans to Fe3+. This reaction precipitated prodigious amounts of ferric oxides, leading to banded iron formations in sedimentary rocks. More importantly, it led to a very slow accumulation of oxygen in the atmosphere. Oxygen was toxic to all forms of life, but the gradual increase gave time for cellular respiration to evolve in bacteria.
Hydroxyl radical ( .OH )
If we somehow developed a ratio of a chemical’s importance to how well-known it is to a scientifically literate audience, the hydroxyl radical (.OH ) would probably win the prize. A popular freshman chemistry textbook in North America does not even list hydroxyl as a topic in its index. Yet when it comes to cleaning up after impurities in the atmosphere, it is the most important oxidizing species in the air.
Two molecules of the so-called detergent of the atmosphere are formed for every oxygen radical that reacts with a water molecule. When the pollutant nitrogen dioxide is removed from the atmosphere it is the hydroxyl radical that turns into nitric acid. Similarly, it converts sulfur dioxide pollution into sulfuric acid, although the formation of acids isn’t without consequences.
But there are very few hydroxyl radicals in the air for two reasons. Most oxygen radicals formed by ozone’s ultraviolet dissociation go on to form new ozone. Only 3% combine with water to form hydroxyl. The other reason is that they are short-lived due to their high reactivity, existing for only 0.01 to 1 second after they are formed. This makes hydroxyl radicals extremely difficult to measure. Their tracking must be indirect, usually based on measuring atmospheric chemicals whose sources are well-known and initiated mostly by a reaction with . Even when their concentration is boosted by the tropics’ humidity, more intense sunlight and higher temperatures, their concentration rarely exceeds 2 parts per trillion(ppt) at sea level. Elsewhere on Earth it can be as low as 0.01 ppt (Figure 7).

When we release methane into the atmosphere, hydroxyl radicals turn it into water and a methyl radical which goes on to form formaldehyde. Unfortunately, every molecule of methane, a greenhouse gas, remains in the atmosphere for about 10 years before it’s cleansed by hydroxyl. Luckily, the poison carbon monoxide’s residence time is only 3 months. But upon attack by hydroxyl, it becomes the greenhouse gas carbon dioxide.
In short, we must be thankful for hydroxyl’s existence. The air quality would certainly be worse without it. But because of its slow kinetics and some undesirable products, it is not the solution to our pollution. When carbon monoxide is present, it consumes about 70% of hydroxyl in its immediate environment. Much of the other 30% attacks methane, which regenerates carbon monoxide. In Chemistry of Atmospheres, Richard Wayne pointed out that since an average adult male ingests a total of 3.2 kg of food and water but inhales 13.5 kg of air per day, the air quality is at least as important as the cleanliness of food and water.This includes indoor air quality, and unfortunately hydroxyl’s role in that context has received far less attention. Due to the radical’s short lifetime, it cannot be assumed to be transferred from outside air. But it does exist inside homes. In 2013, Gligorovski directly measured concentrations of 1.8 × 106molecules per cm3. It forms when short wavelengths of light breakdown HONO, a gas derived from NO2, which in turn forms from cigarette smoke, fireplaces, furnaces, car exhaust (from outside or a garage) and from gas stoves.
Advanced oxidation processes(AOP) are being developed to treat municipal and industrial waste take. These techniques take advantage of hydroxyl radicals. They are usually generated by coupled chemical and/or physical systems including combinations of hydrogen peroxide (H2O2) with ferrous ion (Fe2+)ferric ion (Fe3+) catalyst and ozone. To increase the concentration of the radical, ultraviolet lamps are used. Analyzing the intermediate reactions and products is a challenging task. Many occur in less than 100 microseconds. Research into AOP’s chemistry, not the technique itself, relies on a combination of pulse radiolysis (which uses highly accelerated electrons from a linear accelerator) and competition kinetics methods, laser-induced fluorescence, and electron spin resonance (ESR). The latter resembles proton nuclear magnetic resonance(1H NMR), but it excites the spins of electron instead of those of protons. ESR spectroscopy (Figure 8) was developed to study radicals such as hydroxyl. Medical research also makes use of ESR to examine the link between some diseases and oxidative damage by radicals such as .OH.